天然产物Caulamidine D和 (−)-Isocaulamidine D的全合成
天然产物卤化吲哚类生物碱Caulamidines B–D (1, 3, 5)和isocaulamidines B–D (2, 4, 6) (图 1)是由美国科学家Gustafson及其同事于2023年从海鞘Polyandrocarpa sp.的提取物中分离获得 的(图 2)[1],可作为潜在的抗疟原虫药物先导物。

图 1天然产物Caulamidines B–D和isocaulamidines B–D
(图片来源于J. Am. Chem. Soc)
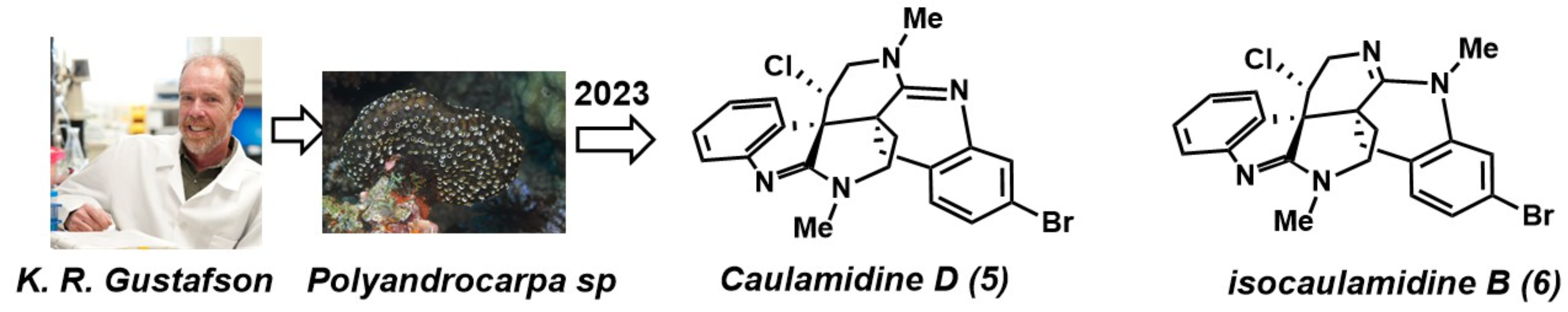
图2 天然产物Caulamidines D (5)和isocaulamidines D (6)的分离
(图片来源于百度)
天然产物(−)-caulamidine D (5)和(−)-isocaulamidine D (6)均含有六氢-2,6-naphthyridine以及二氢吲哚衍生的四氢喹啉相互稠合的并环系统[2-3]、C10和C23具有邻近的季碳立体中心、C11处手性氯原子和C18位溴原子。
中国海洋大学的徐涛教授团队在J. Am. Chem. Soc上,以 “Enantioselective Total Syntheses of (−)-Caulamidine D and (−)-Isocaulamidine D and Their Absolute Configuration Reassignment” 为题,首次报道了天然产物 (−)-caulamidine D和 (−)-isocaulamidine D的对映选择性全合成,并通过X射线晶体学明确地阐明了二者的绝对构型。关键步骤主要包括:不对称Meerwein-Eschenmoser-Claisen重排来构建具有挑战性的C10, C23连续立体中心(Meerwein–Eschenmoser–Claisen rearrangement); 6-exo-dig/6-exo-tet胺/腈环化反应(6-exo-dig/6-exo-tet amine/nitrile cyclization reaction.)。
其全合成步骤如下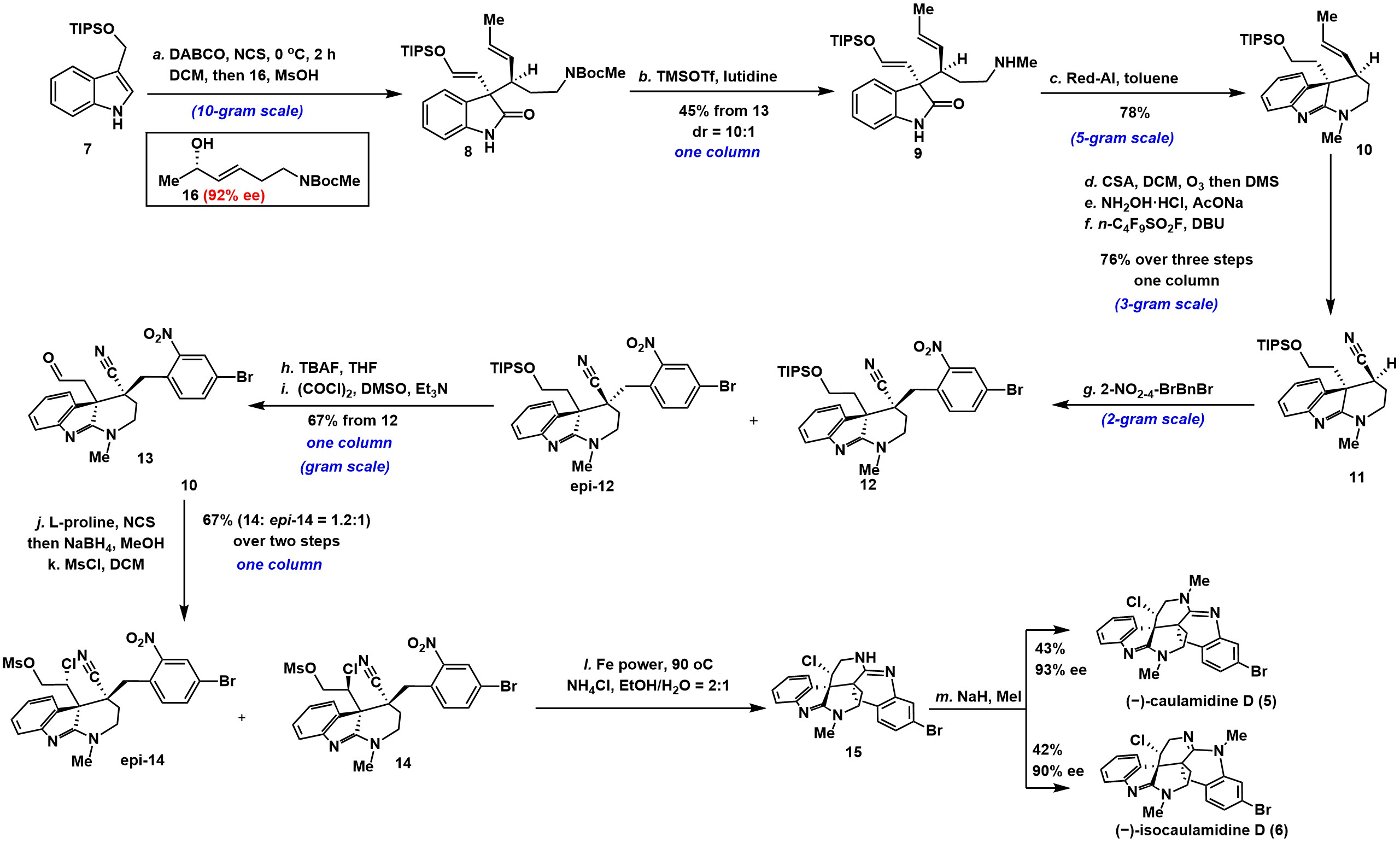
图3 (−)-caulamidine D (5)和(−)-isocaulamidine D (6)全合成
(图片来源于J. Am. Chem. Soc)
采用TBS保护的3-吲哚乙醇(7)和手性烯丙醇(16)为原料,通过Meerwein-Eschenmoser-Claisen重排[2-3]获得邻位具有立体中心获得产物(8),(8)脱除Boc保护基获得砌块(9),而后(9) 在Re-Al体系下构建哌啶环骨架获得(10)。随后,(10)经臭氧化、醛和羟胺缩合和消除反应共三步获得腈(11),而后(11)在LDA作用下发生SN2取代反应获得(12)和(epi-12),(12)经TBAF脱除TIPS保护基以及Swern 氧化两步获得醛中间体(13)。之后(13)经亲电氯代、还原和Ms保护三步获得(14)和(epi-14),(14)经6-exo-dig/6-exo-tet胺/腈环化反应获得关键的中间体(15)。最后,(15)在碱的作用下发生N-甲基化反应完成天然产物(−)-caulamidine D (5)和(−)-isocaulamidine D (6)的全合成。并通过X射线晶体学明确地阐明了它们的绝对构型。此外,用TFA对的−)-isocaulamidine D的滴定实验表明,当TFA被添加到(6)的CD3OD溶液中时,观察到明显的信号偏移,如 C11和C22质子。当加入0.7μL TFA时,1H和13C NMR数据都与报道的NMR数据非常一致。
原文:Enantioselective Total Syntheses of (−)-Caulamidine D and (−)-Isocaulamidine D and Their Absolute Configuration Reassignment Haiyong Yu, Junhao Zhang, Dongxu Ma, Xiaotong Li, and Tao Xu* J. Am. Chem. Soc., 2023, ASAP. https://pubs.acs.org/doi/10.1021/jacs.3c08714.
参考文献
- X. Tian, D. Wang, W. Jiang, H. R. Bokesch, B. A. P. Wilson, B. R. O’Keefe, K. R. Gustafson, J. Nat. Prod. 2023, 86, 1855. doi:10.1021/acs.jnatprod.3c00393.
- M. Wang, M. Feng, B. Tang, X. Jiang, Tetrahedron Lett. 2014, 55, 7147. doi:10.1016/j.tetlet.2014.10.152.
- A. Steven, L. E. Overman, Angew. Chem. Int. Ed. 2007, 46, 5488. doi: 10.1002/anie.200700612.
相关化合物
| 品名 | CAS | 货号 |
|---|---|---|
| Trifluoroacetic acid, 99% 三氟乙酸 , 99% | 76-05-1 | 252705 |
| 3-(2-Hydroxyethyl)indole 3-(2-羟乙基)吲哚 , >97% | 526-55-6 | SY004666 |
| Tetrabutylammonium fluoride, 75 wt.% solution in H2O 四丁基氟化铵 , 75 wt.% 水溶液 | 429-41-4 | 915029 |
| 1,4-Diazabicyclo[2.2.2]octane, 97% 1,4-二叠氮双环[2.2.2]辛烷 , 97% | 280-57-9 | 129882 |
| Trimethylsilyl trifluoromethanesulfonate, 99% 三氟甲烷磺酸三甲基硅酯 , 99% | 27607-77-8 | 264281 |
| (+)-10-Camphorsulfonic acid, 99%, reagent grade (+)-10-樟脑磺酸 , 99% , 试剂级 | 3144-16-9 | 989023 |
相关阅读
七大系列,上万个产品,助力天然产物全合成研究!天然产物化学——助力提取、修饰、全合成、药理与代谢研究
光延反应的关键中间体——DEAD/DIAD,用于天然产物全合成
服务科技与工业发展 造福人类

关注微信公众号

